Results
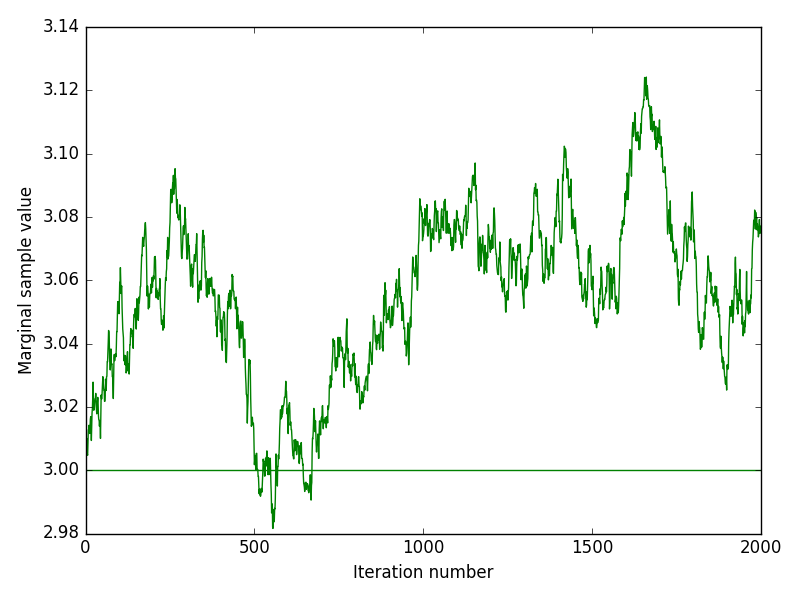
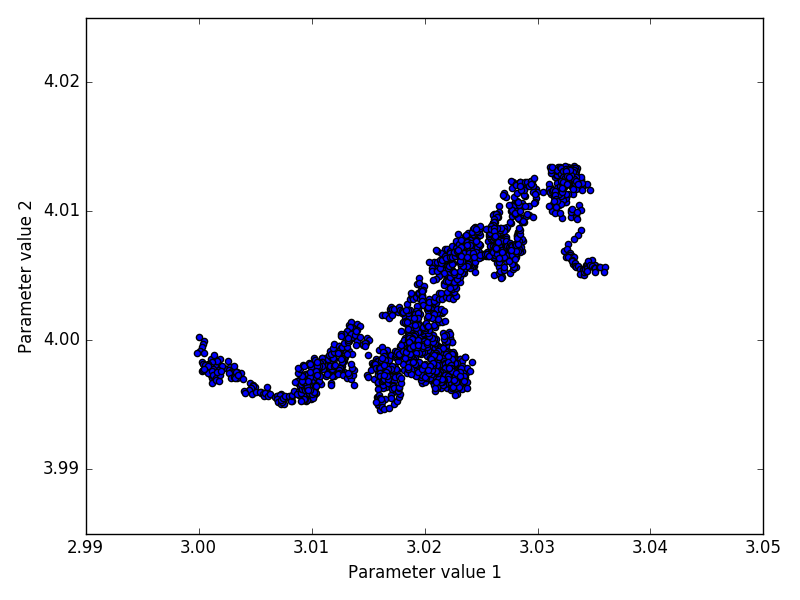
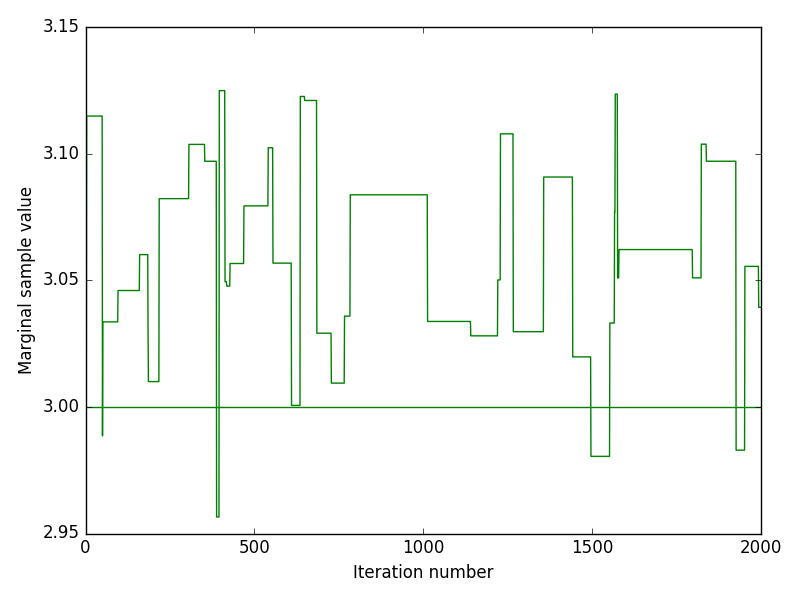
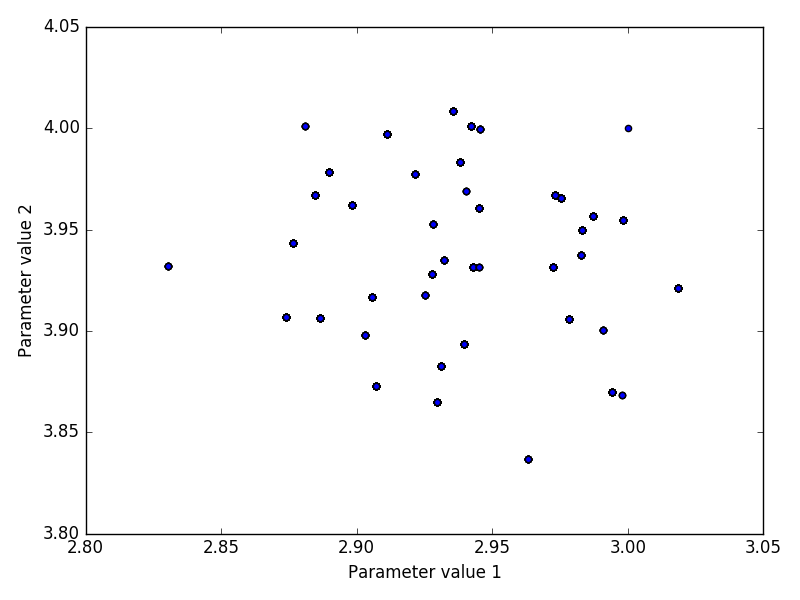
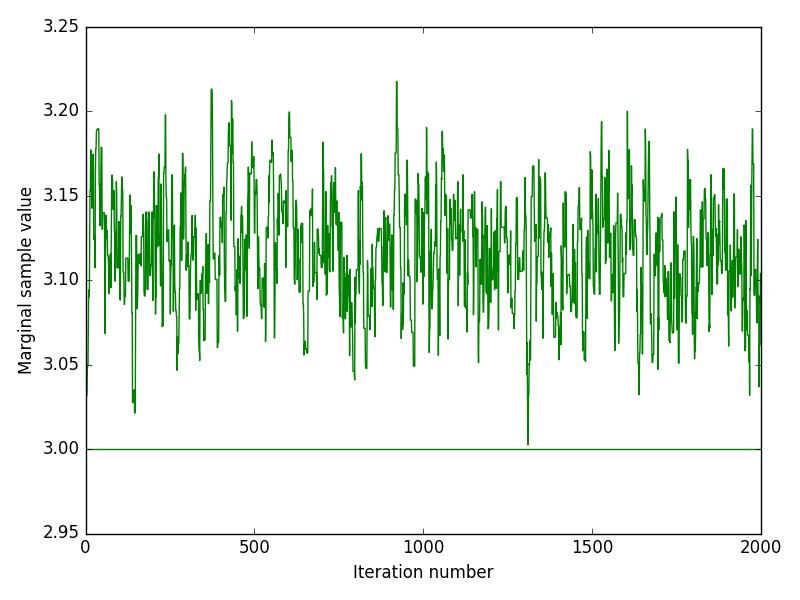
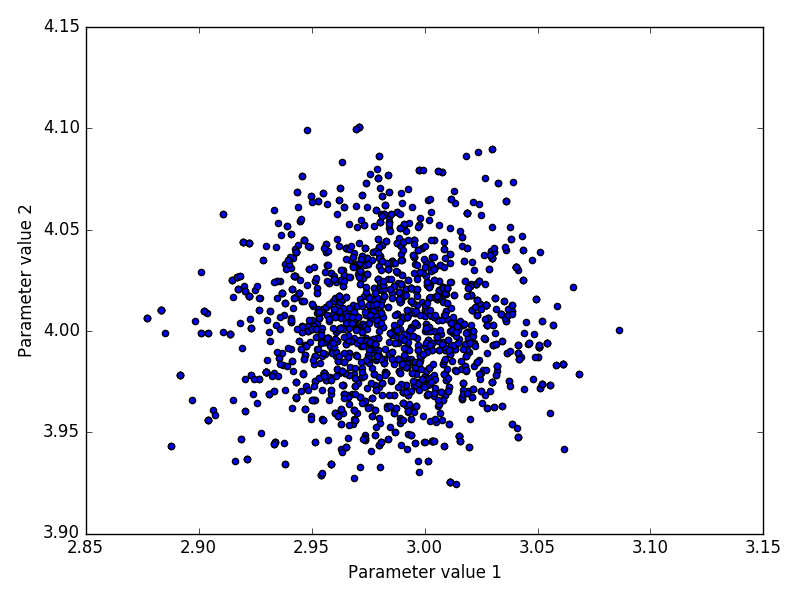
The inference method was performed on both experimental and in-silico simulated data. Below we discuss the performance of the inference by looking at indicative plots such as joint and marginal posteriors, trace plots or Bayesian credible intervals.
Experimental data
The experimental data consists of qPCR amplification curves in the form of arrays of fluorescence reads. Each amplification curve represents the observation data used to infer the initial quantity of a target analyte X0 (number of mtDNA molecules in a single cell). The qPCR experiments are performed in parallel, with the contents of each target analyte being placed in a separate well.
Each amplification curve from experimental data has a corresponding X0 estimate obtained using the standard curve method. This value is used as an additional indicator for whether the inferred posterior distribution for X0 is centered around to the true value of the parameter. However, this value obtained from the standard curve method is simply an estimate, and should not be treated as the true X0.
In-silico simulation data
We perform in-silico simulations to obtain an amplification curve given a parametrisation θ = [X0, r, σ], a known fluorescence coefficient α (which is not inferred) and a number of qPCR cycles n. The simulated process starts with X=X0 molecules, and is updated by sampling from a Binomial distribution with probability r. The fluorescence reads F1:n are generated by adding noise sampled from a Gaussian distribution with variance σ (see Model).
The advantage of the in-silico data is that it allows us to compare the means and posterior distributions of all parameters X0, r, σ with the true values using simulated data F1:n.